Innehåll
- orsaker
- symtom
- Tentor och prov
- Behandling
- Utsikter (prognos)
- När ska du kontakta en medicinsk professionell
- Förebyggande
- Alternativa namn
- Bilder
- referenser
- Recension Datum 8/7/2017
Creutzfeldt-Jakobs sjukdom (CJD) är en form av hjärnskada som leder till en snabb minskning av rörelse och mental funktion.
orsaker
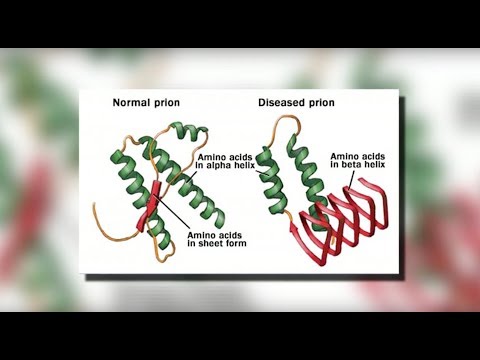
CJD orsakas av ett protein som kallas prion. Ett prion får normala proteiner att vikas onormalt. Detta påverkar andra proteins förmåga att fungera.
CJD är mycket sällsynt. Det finns flera typer. De klassiska typerna av CJD är:
- Sporadisk CJD utgör de flesta fallen. Det förekommer utan känd anledning. Den genomsnittliga ålder vid vilken den börjar är 65.
- Familial CJD uppstår när en person ärver den onormala prion från en förälder (denna form av CJD är sällsynt).
- Förvärvat CJD innehåller variant CJD (vCJD), formen relaterad till galna ko sjukdomar. Iatrogen CJD är också en förvärvad form av sjukdomen. Iatrogen CJD passerar ibland genom blodtransfusion, transplantation eller förorenade kirurgiska instrument.
Variant CJD orsakas av att äta smittat kött. Infektionen som orsakar sjukdomen hos kor tros vara den samma som orsakar vCJD hos människor.
Variant CJD orsakar mindre än 1% av alla CJD-fall. Det tenderar att påverka yngre människor. Färre än 200 personer världen över har haft denna sjukdom. Nästan alla fall inträffade i England och Frankrike.
CJD kan vara relaterad till flera andra sjukdomar orsakade av prioner, inklusive:
- Kronisk slösande sjukdom (hittad i hjort)
- Kuru (drabbade mestadels kvinnor i Nya Guinea som åt hjärnor av döda släktingar som en del av en begravningsritual)
- Scrapie (finns hos får)
- Andra mycket sällsynta ärftliga mänskliga sjukdomar, såsom Gerstmann-Straussler-Scheinker-sjukdomen och dödlig familjär sömnlöshet
symtom
CJD-symtom kan innehålla något av följande:
- Demens som förvärras snabbt över några veckor eller månader
- Suddig syn (ibland)
- Gångförändringar (gång)
- Förvirring, desorientering
- Hallucinationer (se eller höra saker som inte finns där)
- Bristande samordning (till exempel stumbling och fallande)
- Muskelstyvhet, rubbning
- Känsla nervös, hoppig
- Personlighetens förändringar
- sömnighet
- Plötsliga ryckiga rörelser eller anfall
- Problemlösning
Tentor och prov
Tidigt i sjukdomen kommer ett nervsystem och mental undersökning att visa minne och tänkande problem. Senare i sjukdomen kan en motorsystemsundersökning (en prov för att testa muskelreflexer, styrka, koordination och andra fysiska funktioner) visa:
- Onormala reflexer eller ökade normala reflexsvar
- Ökning av muskelton
- Muskelsprängning och spasmer
- Starkt startande svar
- Svaghet och förlust av muskelvävnad (muskelavfall)
Det finns en förlust av koordination och förändringar i cerebellum. Det här är hjärnans område som styr samordningen.
En ögonprov visar områden av blindhet som personen kanske inte märker.
Test som används för att diagnostisera detta tillstånd kan innefatta:
- Blodprov för att utesluta andra former av demens och att leta efter markörer som ibland uppstår med sjukdomen
- CT-skanning av hjärnan
- Elektroencefalogram (EEG)
- MR i hjärnan
- Spinal tap för att testa för ett protein som kallas 14-3-3
Sjukdomen kan bara bekräftas med hjärnbiopsi eller obduktion. Idag är det väldigt sällsynt att en hjärnbiopsi görs för att leta efter denna sjukdom.
Behandling
Det finns ingen känd botemedel för detta tillstånd. Olika läkemedel har försökt att sakta ner sjukdomen. Dessa inkluderar antibiotika, läkemedel mot epilepsi, blodförtunnare, antidepressiva medel och interferon. Men ingen fungerar bra.
Målet med behandlingen är att tillhandahålla en säker miljö, kontrollera aggressivt eller agitat beteende och möta personens behov. Det kan kräva övervakning och hjälp i hemmet eller på vårdcentralen. Familjrådgivning kan hjälpa familjen att klara de förändringar som behövs för hemvård.
Människor med detta tillstånd kan behöva hjälp med att kontrollera oacceptabelt eller farligt beteende. Detta innebär att belöna positiva beteenden och ignorera negativa beteenden (när det är säkert). De kan också behöva hjälp att bli orienterad mot omgivningen. Ibland behövs läkemedel för att hjälpa till att kontrollera aggression.
Personer med CJD och deras familj kan behöva söka juridisk rådgivning tidigt under sjukdomen. Advance direktiv, fullmakt och andra rättsliga åtgärder kan göra det lättare att fatta beslut om vården av personen med CJD.
Utsikter (prognos)
Resultatet av CJD är mycket dåligt.Personer med sporadisk CJD kan inte ta hand om sig själv inom 6 månader eller mindre efter att symtomen börjar.
Störningen är dödlig på kort tid, vanligtvis inom 8 månader. Människor som har variant CJD blir värre långsammare, men tillståndet är fortfarande dödligt. Några personer överlever så länge som 1 eller 2 år. Dödsorsaken är vanligtvis infektion, hjärtsvikt eller andningssvikt.
Kursen av CJD är:
- Infektion med sjukdomen
- Förlust av förmåga att interagera med andra
- Förlust av förmåga att fungera eller ta hand om sig själv
- Död
När ska du kontakta en medicinsk professionell
CJD är inte en medicinsk nödsituation. Att bli diagnostiserad och behandlad tidigt kan dock göra symptomen lättare att kontrollera, ge patienterna tid att göra förhandsdirektiv och förbereda sig för livets slut och ge familjer extra tid att komma överens med tillståndet.
Förebyggande
Medicinsk utrustning som kan vara förorenad bör tas bort från service och bortskaffas. Människor som är kända för att ha CJD borde inte donera ett hornhinna eller annan kroppsvävnad.
De flesta länder har nu strikta riktlinjer för hantering av smittade kor för att undvika att överföra CJD till människor.
Alternativa namn
Överförbar spongiform encefalopati; vCJD; CJD; Jacob-Creutzfeldt sjukdom
Bilder

Creutzfeldt-Jakob sjukdom
Central nervsystemet och perifert nervsystem
referenser
Bosque PJ, Tyler KL. Prions- och prionsjukdomar i centrala nervsystemet (transmissibla neurodegenerativa sjukdomar). I: Bennett JE, Dolin R, Blaser MJ, eds. Mandell, Douglas och Bennetts principer och praxis för smittsamma sjukdomar, uppdaterad utgåva. 8: e upplagan Philadelphia, PA: Elsevier Saunders; 2015: chap 181.
Geschwind MD. Prionsjukdomar. I: Daroff RB, Jankovic J, Mazziotta JC, Pomeroy SL, eds. Bradleys neurologi i klinisk praxis. 7: e upplagan Philadelphia, PA: Elsevier; 2016: kap 94.
Recension Datum 8/7/2017
Uppdaterad av: Amit M. Shelat, DO, FACP, Att delta neurolog och biträdande professor i klinisk neurologi, SUNY Stony Brook, School of Medicine, Stony Brook, NY. Granskning tillhandahållen av VeriMed Healthcare Network. Också granskad av David Zieve, MD, MHA, medicinsk chef, Brenda Conaway, redaktionschef och A.D.A.M. Redaktionellt lag.