
Innehåll
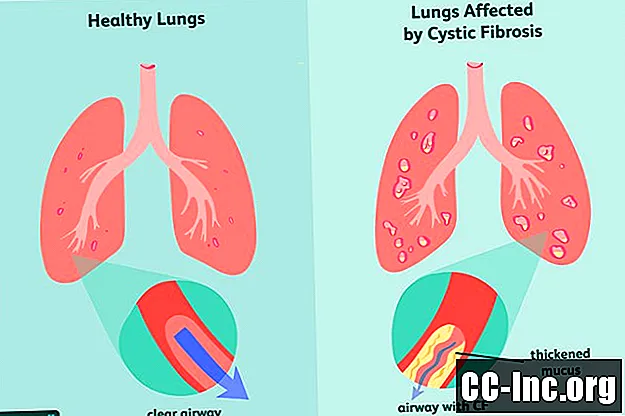
Cystisk fibros (CF) är en ärftlig, livshotande sjukdom som skadar lungorna och mag-tarmkanalen. Det orsakas av en defekt gen som utlöser produktionen av förtjockat slem som täpper till luftvägarna och blockerar utsöndringen av matsmältningsenzymer.Symtomen är progressiva och ofta svåra, och de kan inkludera andningsproblem, återkommande lunginfektioner, dålig tillväxt, manlig infertilitet och kronisk inflammation i bukspottkörteln, levern, njurarna och hjärtat.
CF kan diagnostiseras med blodprov, genetisk screening och ett förfarande som kallas ett svettkloridtest.
Även om det inte finns något botemedel mot CF, finns det behandlingar som kan förbättra både livslängden och kvaliteten på ens liv.
Dessa inkluderar luftvägsklareringstekniker, inhalerade antibiotika, slemförtunnande medel, bukspottkörtelnzymer, en kaloririk diet och nyare läkemedel som kallas CFTR-modulatorer. I svåra fall kan en lungtransplantation behövas.
Cystisk fibros Symtom
Som en genetisk störning är cystisk fibros något du är född med. Det kan eller inte kan ha symtom vid tidpunkten för födseln och kan ofta ta månader eller till och med år innan några tecken på sjukdom uppträder. Vid den tiden kan lungorna och mag-tarmkanalen redan ha upplevt skador som inte kan ångras.
De vanligaste tidiga tecknen och symtomen på CF inkluderar:
- Blockering av barnets första avföring (mekonium)
- Salt smakande hud
- En kronisk hosta, väsande andning eller färgad sputum
- Lös, fet och typiskt illaluktande avföring
- Lunginfektion, ofta återkommande
- Dålig tillväxt och misslyckande med att trivas
Om inte dessa symtom kan kontrolleras kan stress på lungorna (och oförmågan att gå upp i vikt) ha en kumulativ effekt, vilket påverkar flera organ och ökar risken för sjukdomskomplikation.
Några av de mer karakteristiska komplikationerna inkluderar:
- Försenad pubertet
- Bronkiektas (kronisk förtjockning av lungväggarna)
- Viktminskning
- Pankreatit (inflammation i bukspottkörteln)
- Manlig infertilitet
- Pulmonell hypertoni (högt blodtryck i lungan)
- Gallstenar
- Cystisk fibrosrelaterad diabetes
- Cor pulmonale (höger hjärtsvikt)
- Cirros (funktionell ärrbildning i levern)
Eftersom CF orsakar progressiv skada på celler och vävnader kommer alla skador som orsakas i lungorna och andra organ i stort sett att vara irreversibla. Döden är oftast resultatet av andningssvikt, följt av hjärtsvikt och leversvikt.
Symtom på cystisk fibros
Orsaker
Cystisk fibros orsakas av mutationen av cystisk fibros transmembranreceptor (CFTR) -genen, som är ansvarig för att producera CFTR-proteinet. Detta är det protein som kroppen behöver för att reglera flödet av salt och vatten in och ut ur celler . Om proteinet är deformerat eller defekt kan det orsaka uttorkning på ytan av en cell, vilket leder till förtjockning av det omgivande slem.
CF är en autosomal recessiv sjukdom, vilket innebär att du måste ärva CFTR-mutationen från både din mor och far för att få sjukdomen. Om du bara ärver en defekt gen kommer du inte att ha CF utan istället vara en bärare av den muterade genen.
Du kan ärva sjukdomen om var och en av dina föräldrar antingen har en CFTR-mutation eller CF själv. Om båda föräldrarna är bärare skulle du ha:
- 25 procents chans att få CF
- 50 procents chans att bli transportör
- 25 procents chans att bli opåverkad
Å andra sidan, om en av dina föräldrar är transportör och den andra har CF, har du 50/50 chans att antingen få CF eller vara bärare.
Cystisk fibros är en av de vanligaste genetiska sjukdomarna och drabbar ungefär en av varje 2500 barn födda i USA.
Det är vanligast bland kaukasiska och latinamerikaner och förekommer mindre ofta hos människor av afrikansk eller asiatisk härkomst.
Riskfaktorer för cystisk fibrosDiagnos
Det finns några test som används för att diagnostisera cystisk fibros. De fungerar antingen genom att direkt upptäcka CFTR-mutationen eller indirekt mäta biologiska förändringar som överensstämmer med sjukdomen. Diagnosmetoden kan variera under graviditeten, när barnet föds eller när som helst därefter.
Diskussionsguide för cystisk fibros doktor
Få vår utskrivbara guide för din nästa läkarmöte för att hjälpa dig att ställa rätt frågor.

Av de två standardtester som vanligtvis används för att diagnostisera CF:
- Svettklorid testning, även känt som svettest, mäter mängden klorid på huden. Eftersom CF stör saltöverföringen till och från celler, kommer det att ackumuleras salt i svett.
- Genetisk CFTR-testning används för att detektera de vanligaste mutationerna av CFTR-mutationen. Medan det finns över 2000 CFTR-mutationer kända för att orsaka cystisk fibros, representerar de 23 som ingår i standardpanelen de mest troliga misstänkta.
Under graviditeten kan CFTR-genetiska testet användas för att testa vätskor som erhållits genom fostervattensprov eller celler som extraherats via provtagning av korionvillor (CVS).
Nyfödda screening används också standard för att diagnostisera CF och är idag mandat i alla 50 stater och District of Columbia. Vad detta innebär kommer att skilja sig beroende på var i USA du bor. Om de nyfödda screeningresultaten är positiva skulle ett svettest användas för att bekräfta diagnosen.
Hur diagnostiseras cystisk fibrosBehandling
Även om det inte finns något botemedel mot cystisk fibros har framstegen i behandlingen förlängt livslängden för dem som lever med sjukdomen.
Målet med CF-behandling är fyrfaldigt: att förhindra infektioner, bibehålla lungfunktionen, normalisera matsmältningen och sakta sjukdomsprogressionen.
Bland de terapeutiska verktygen som används för att hantera CF:
- Luftvägarens tekniker (ACT) utförs för att lossa och driva ackumulerat slem från lungorna. Teknik inkluderar huff hosta, bröstverkan eller svängning i bröstväggen.
- En fettrik kost med hög kaloriinnehåll används för att kompensera för malabsorption av fetter, proteiner och näringsämnen i tarmarna.
- Pankreasenzymtillskott används för att stärka matsmältningsenzymerna som bukspottkörteln inte kan producera på grund av överdriven ansamling av slem.
- Antibiotika tas dagligen för att förhindra bakteriella lunginfektioner.
- Mukolytika-droger som används för att tunna slem före ACT-kan användas.
- CFTR-modulatorer är en ny klass av läkemedel som kan korrigera vissa defekter i CFTR-proteinet och återställa deras reglerande funktion.
- Syrebehandling kan användas under akuta episoder när din andning har blivit kraftigt nedsatt.
- Enteral näring, även känd som tubmatning, kan användas om du inte kan hålla i vikt genom normal näring.
- Lungtransplantation övervägs när dina lungor inte längre kan överleva utan mekanisk ventilation.
Hantera
År 1938, när cystisk fibros först klassificerades som en sjukdom, levde barn sällan längre än sitt första levnadsår. Vid 1980-talet kunde man förvänta sig att leva så länge 20 till 25 år. Idag har bilden förändrats helt och hållet med människor som bor långt in i 40- och till och med 50-talet om behandlingen påbörjas tidigt och följs.
Detta betyder inte att CF är mindre allvarlig än någonsin. Det är en livsförändrande händelse som kräver flit och konsistens för att inte bara klara av sjukdomen utan leva högsta möjliga livsstandard.
För detta ändamål måste du normalisera CF i ditt liv genom att fastställa rutiner och metoder för att undvika upp-och nedgångar som kan orsaka stress och öka funktionshinder. Bland övervägandena måste du:
- Hantera din näring. Människor med CF behöver ofta dubbelt så många kalorier dagligen som andra gör.
- Träna regelbundet. Fitnessrutiner bör helst involvera minst 20 till 30 minuters aerob aktivitet tre gånger i veckan. Hitta något roligt du kan göra under en livstid.
- Håll väl hydrerad. Detta gör att lungorna och tarmarna fungerar ordentligt. Beroende på din ålder bör du dricka inte mindre än sex till åtta höga glas vatten per dag.
- Utför luftvägsavstånd korrekt. När dina hälsobehov förändras kan också de typer av rensningsverktyg du behöver. Tala med din pulmonolog eller sjukgymnast om du inte uppnår de resultat du borde.
- Sök support. Förutom vänner och familj kan du kontakta närmaste kapitel i Cystic Fibrosis Foundation (CFF) för att ansluta till ett supportnätverk i ditt område.
- Sök ekonomisk hjälp. CFF erbjuder tjänster som hjälper familjer att bättre hantera de höga kostnaderna för CF-behandling.
Ett ord från Verywell
Medan nyfödda screeningar dramatiskt har ökat frekvensen av CF-diagnoser hos spädbarn, görs över 25 procent av diagnoser endast under barndomen, tonåren och de tidiga vuxna åren.
Detta är problematiskt eftersom tidig diagnos och behandling kan avvärja många av de allvarligare komplikationerna av CF innan någon allvarlig skada kan göras. Medan behandlingen inte kan stoppa eller vända sjukdomen, kan det säkerställa många fler sjukdomsfria år.
För detta ändamål är det viktigt att känna till de tidiga symptomen på CF och att prata med din läkare om du misstänker att ditt barn kan ha sjukdomen. Detta gäller särskilt i stater som skärmar med endast IRT-blodprov, vilket kan leda till att så många 5 procent av barn upplever antingen en fördröjd diagnos eller ett falskt negativt resultat, enligt forskning från University of Wisconsin School of Medicine and Public Health. .
Vilka symtom kan du förvänta dig med cystisk fibros?