
Innehåll
- orsaker
- symtom
- Tentor och prov
- Behandling
- Supportgrupper
- Utsikter (prognos)
- När ska du kontakta en medicinsk professionell
- Förebyggande
- Alternativa namn
- Bilder
- referenser
- Recension Datum 10/15/2018
Niemann-Pick-sjukdomen (NPD) är en grupp av sjukdomar som går ner genom familjer (ärftliga), där fettämnen som kallas lipider samlar i mjälten, lever och hjärnans celler.
Det finns tre vanliga former av sjukdomen:
- Typ A
- Typ B
- Typ C
Varje typ involverar olika organ. Det kan eller kanske inte involvera nervsystemet och andas. Var och en kan orsaka olika symtom och kan uppträda vid olika tidpunkter i livet.
orsaker
NPD-typerna A och B uppträder när celler i kroppen inte har något enzym som kallas surt sphingomyelinas (ASM). Detta ämne hjälper till att bryta ner (metabolisera) ett fettämne som kallas sphingomyelin, vilket finns i varje cell i kroppen.
Om ASM saknas eller inte fungerar ordentligt, byggs sphingomyelin upp i cellerna. Detta dödar dina celler och gör det svårt för organen att fungera ordentligt.
Typ A förekommer i alla raser och etniciteter. Det är vanligare i Ashkenazi (östeuropeiska) judiska befolkningen.
Typ C uppstår när kroppen inte kan bryta ner kolesterol och andra fetter (lipider). Detta leder till för mycket kolesterol i levern och mjälte och för mycket av andra lipider i hjärnan. Typ C är vanligast bland Puerto Ricans spanska härkomst.
Typ C1 är en variant av typ C. Det innebär en defekt som stör hur kolesterol rör sig mellan hjärnceller. Denna typ har bara setts i franska kanadensiska folk i Yarmouth County, Nova Scotia.
symtom
Symtom varierar. Andra hälsoförhållanden kan orsaka liknande symtom. De tidiga skeden av sjukdomen kan bara orsaka några symtom. En person får aldrig ha alla symtom.
Typ A börjar vanligtvis under de första månaderna av livet. Symtom kan innehålla:
- Abdominal (mageområde) svullnad inom 3 till 6 månader
- Körsbärsröd fläck på baksidan av ögat (på näthinnan)
- Feeding svårigheter
- Förlust av tidiga motoriska färdigheter (blir sämre över tiden)
Typ B-symtom är vanligtvis mildare. De förekommer i sen barndom eller tonåren. Buksvullnad kan förekomma hos små barn. Det finns nästan inget hjärnans och nervsystemets engagemang, till exempel förlust av motoriska färdigheter. Vissa barn kan ha upprepade andningsinfektioner.
Typer C och C1 påverkar vanligtvis barn i skolåldern. Det kan dock uppstå vilken tid som helst mellan tidig spädbarn och vuxen ålder. Symtom kan innehålla:
- Svårighetsgrad rörliga lemmar som kan leda till ostabil gång, klumpighet, gångproblem
- Förstorad mjälte
- Förstorad lever
- Gulsot vid (eller kort efter) födelse
- Inlärningssvårigheter och intellektuell nedgång
- kramper
- Släckt, oregelbundet tal
- Plötslig förlust av muskelton som kan leda till fall
- Tremors
- Problem att flytta ögonen upp och ner
Tentor och prov
Ett blod- eller benmärgstest kan göras för att diagnostisera typ A och B. Testet kan berätta vem som har sjukdomen, men visar inte om du är bärare. DNA-test kan göras för att diagnostisera bärare av typ A och B.
En hudbiopsi görs vanligtvis för att diagnostisera typerna C och D. Hälso- och sjukvårdsleverantören tittar på hur hudcellerna växer, flyttar och lagrar kolesterol. DNA-test kan också göras för att leta efter de 2 gener som orsakar denna typ av sjukdomen.
Andra tester kan innefatta:
- Benmärgs aspiration
- Leverbiopsi (vanligtvis inte behövs)
- Slit-lampa ögonprov
- Test för att kontrollera nivå av ASM
Behandling
Vid denna tidpunkt finns ingen effektiv behandling för typ A.
Benmärgstransplantationer kan försökas för typ B. Forskare fortsätter att studera möjliga behandlingar, inklusive enzymbyte och genterapi.
Ett nytt läkemedel som heter miglustat är tillgängligt för nervsystemet symtom av typ C.
Högt kolesterol kan hanteras med en hälsosam kost med lågt kolesterolvärde eller läkemedel. Forskningen visar emellertid inte att dessa metoder hindrar sjukdomen från att förvärras eller förändra hur cellerna bryter ner kolesterol. Läkemedel finns tillgängliga för att kontrollera eller lindra många symptom, till exempel plötslig förlust av muskelton och anfall.
Supportgrupper
Dessa organisationer kan ge stöd och mer information om Niemann-Pick-sjukdomen:
- National Institute of Neurological Disorders and Stroke - www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- Nationell Niemann-Pick sjukdomsstiftelsen - nnpdf.org
- Nationell organisation för sällsynta sjukdomar - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Utsikter (prognos)
NPD typ A är en allvarlig sjukdom. Det leder vanligtvis till döden efter 2 eller 3 års ålder.
De med typ B kan leva till sen barndom eller vuxen ålder.
Ett barn som visar tecken på typ C före ålder 1 kan inte leva i skolåldern. De som uppvisar symtom efter att ha gått i skolan kan leva i sina mid-late teens. Vissa kan leva in i 20-talet.
När ska du kontakta en medicinsk professionell
Gör ett avtal med din leverantör om du har en familjhistoria för Niemann-Pick-sjukdomen och du planerar att få barn. Genetisk rådgivning och screening rekommenderas.
Ring din leverantör om ditt barn har symtom på denna sjukdom, inklusive:
- Utvecklingsproblem
- Matningsproblem
- Dålig viktökning
Förebyggande
Alla typer av Niemann-Pick är autosomala recessiva. Det innebär att båda föräldrarna är bärare. Varje förälder har 1 kopia av den onormala genen utan att ha några tecken på sjukdomen själva.
När båda föräldrarna är bärare, finns det 25% chans att deras barn kommer att få sjukdomen och 50% chans att deras barn kommer att vara bärare.
Test av bärare detektering är endast möjligt om den genetiska defekten identifieras. Felaktigheterna i typ A och B har undersökts väl. DNA-tester för dessa former av Niemann-Pick är tillgängliga.
Genetiska defekter har identifierats i DNA hos många personer med typ C. Det kan vara möjligt att diagnostisera personer som bär den onormala genen.
Några centra erbjuder test för att diagnostisera en baby som fortfarande är i livmodern.
Alternativa namn
NPD; Sfingomyelinasbrist; Lipidlagerstörning - Niemann-Pick-sjukdom; Lysosomal lagringssjukdom - Niemann-Pick
Bilder
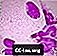
Niemann-Pick skumdjursceller
referenser
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Defekter i metabolism av lipider. I: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20: e upplagan. Philadelphia, PA: Elsevier; 2016: chap 86.
Patterson MC, Hendriksz CJ; NP-C riktlinjer arbetsgrupp, et al. Rekommendationer för diagnos och hantering av Niemann-Pick sjukdom typ C: en uppdatering. Mol Genet Metab. 2012; 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Inborna metabolismfel. I: Turnpenny PD, Ellard S, eds. Emery Elements of Medical Genetics. 15: e utgåvan. Philadelphia, PA: Elsevier; 2017: kap 18
Recension Datum 10/15/2018
Uppdaterad av: Anna C. Edens Hurst, MD, MS, biträdande professor i medicinsk genetik, University of Alabama i Birmingham, Birmingham, AL. Granskning tillhandahållen av VeriMed Healthcare Network. Också granskad av David Zieve, MD, MHA, medicinsk chef, Brenda Conaway, redaktionschef och A.D.A.M. Redaktionellt lag.